
Electronic transitions
In electronic spectroscopy, we excite the electronic system of a molecule. That means that (in first approximation) the movement of the nuclei does not concern us much and we care only about the electrons. In order to excite a transition between different electronic states, the energy of the light has to match the energetic gap between the two electronic states. Additionally, the transition has to be quantum-mechanically allowed. This is governed by the selection rules, which often boil down to a question of symmetry.
In electronic spectroscopy, the excitation has to alter the distribution of electrons within the molecule. In other words, it has to induce a change in the electric dipole moment. This is described by the transition dipole moment 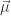

The vector ri describes the movement of each electron i with the elementary charge e. With this, we can compute the intensity of the transition between the initial state 
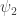
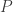
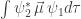
which is called the matrix element. This matrix element has to be non-zero for a transition to be allowed.
Cata-condensed hydrocarbons
Molecules consisting of a chain of benzene rings are called cata-condensed hydrocarbons, and two examples are shown in Figure 1. These molecules are described by the formula C4n+2H2n+4 and each carbon atom belongs to a maximum of two rings. In order to describe the electronic states of these molecules, John Platt introduced a convenient nomenclature based on the free electron orbital method [1]. He postulated that in these systems the electrons travel on a loop of constant potential around the perimeter. In this picture, the energy levels can be described by the simplest model in quantum mechanics, a particle in a one-dimensional box.
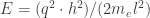
The length of the box l is the perimeter of the hydrocarbon along which the electrons with mass me may travel and q is the so-called ring quantum number. The higher this quantum number, the larger the energy of the molecule. As q gets squared, the energy levels increase quadratically and do not depend on the relative sign of q: electrons with a finite velocity may travel clockwise and counter-clockwise along the perimeter l, giving rise to +q and -q. An exception is the state corresponding to q=0 as in this state the electrons are standing still.

Figure 1: The two cata-condensed hydrocarbons naphthalene (left) and phenanthrene (right). The perimeter determining l is drawn in blue.
Each of the 4n+2 C-atoms contributes a single 

Figure 2: The energy levels of naphthalene in Platt’s theoretical model. The 10 pi-electrons, depicted by blue dots, fill the energy level up to q=2. Hence, q=2 is denoted as f and q=3 as g.
Exciting molecules in cata-condensed hydrocarbons
When we excite the electronic system we lift one electron from the f-shell to the g-shell. The energy of the final state depends on the interaction of the excited electron with the other electrons. We can also be described as the interaction of the excited electron with the hole it created in the f-level. However, we cannot tell whether the electron before (the hole) and after excitation is going clockwise or anti-clockwise as both options have the same energy. As we cannot distinguish between these two options and both contribute, we have to describe the final state by a linear combination.
If an electron is excited from an f(q=n) to a g(q=n+1) shell, the resulting states are described the linear combination: Q=n
La and Lb
Except for highly symmetrical molecules like benzene, the degeneracy in the energy of the states is lifted. The resulting sub-levels are denoted a and b, depending on the orientation of the new nodal plane introduced into the electronic wave function. For the a state, the nodal planes are situated in such a manner, that the electron density is located at the atoms, while the effective charges go to the bonds for the b state. This is illustrated in Figure 3 for the states of naphthalene. According to the third of Hund’s rules, the L states are energetically always lower than the B states.

Figure 3: For naphthalene n=2 and Q equals to 1 for the B-states and 5 for the L-states. Q is also the number of nodal planes. At these planes (dashed lines) no electron density is situated. Hence, the electrons are pushed more to the atoms for the a states and to the bonds for the b states. The resulting orientation of the transition dipole moment is indicated by the arrows.
Compared to naphthalene 2 CH groups are replaced by an NH group in indole. If only the number of
La and Lb in indole
As the molecular symmetry of indole is much lower than the one of benzene, the energetic degeneracy is lifted and we get two separated transitions. In the gas phase, the difference between both states amounts to 2000 cm-1, which is around 5% of the total transition energy. The transition dipole moments of the two states are perpendicular to each other like in the case of naphthalene, shown in Figure 4.

Figure 4: The orientation of the transition dipole moment vectors for the La and Lb state in indole.
Why is this interesting?
The molecule indole is an integral part of the aromatic amino acid tryptophan, essentially it is the reason why tryptophan absorbs UV light. After exciting tryptophan electronically, light is re-emitted. This fluorescence light is widely used to study the structure and dynamics of proteins. The reason for this is the two excited states we just discussed. In an apolar surrounding, the Lb is lowest in energy and hence the emitting state. As soon as the surroundings of tryptophan get more polar, for instance, because the protein changes its structure and water gets in the vicinity of tryptophan, the La is strongly stabilized. The reason for this behaviour lies in the electric dipole moments of the excited states. The one of the La is much larger than the one of the Lb, leading to a favourable dipole-dipole interaction which lowers the energy of the La. Hence, tryptophan can be used as a natural probe to tell us about the structure of the protein or its changes.
Bibliography
[1] J. R. Platt, “Classification of spectra of cata-condensed hydrocarbons” J. Chem. Phys. 17, 484-495 (1948)
[2] G. Weber, “Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds” Biochem. J. 75, 335-345 (1948)
